

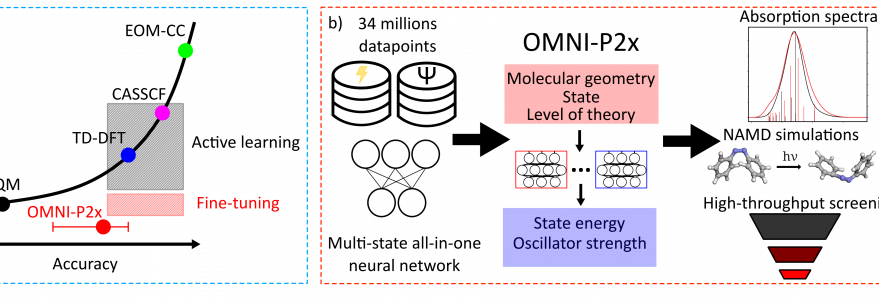
Researchers from the Faculty of Chemistry at the University of Warsaw, in collaboration with scientists from Xiamen University, have developed OMNI-P2x ‒ the first universal neural network capable of predicting how diverse organic molecules behave after absorbing light. The study describing these results has been published in “Nature Communications”.
Photoinduced processes underpin many modern technologies, from solar cells and OLED displays to photocatalysis and solar energy storage. However, modelling such processes at the quantum-mechanical level is computationally demanding. An accurate description of a single molecule may require hours or even days of computing time, significantly limiting the scale of possible studies.
The research was led by Pavlo O. Dral and Joanna Jankowska, while the first author of the publication is Mikołaj Martyka. The team trained the neural network using data for approximately 3.1 million organic molecules. The resulting model, OMNI-P2x, can predict the UV/Vis absorption spectrum of a previously unseen molecule in a fraction of a second, with accuracy comparable to widely used quantum-mechanical methods such as TD-DFT, which require at least several minutes to perform the same task. Importantly, OMNI-P2x proved to be not only faster but also more accurate than popular approximate semi-empirical methods.
AI-assisted design of photoswitches
To demonstrate the practical utility of the model, the authors screened half a million azobenzene derivatives ‒ molecules capable of changing their structure under light irradiation and considered promising candidates for both solar energy storage and medical applications such as photopharmacology. From this vast dataset, the researchers identified 21 top-performing structures, including three with predicted absorption maxima at 640, 667, and 687 nm, surpassing analogous compounds reported previously in the literature.
A second application of the model is the acceleration of simulations of ultrafast excited-state molecular dynamics ‒ processes occurring within the first femtoseconds after photon absorption. By using OMNI-P2x as a starting point for further machine learning, such simulations become up to ten times more data-efficient.
What comes next?
The current version of the model supports molecules composed of the most common organic elements: H, C, N, O, F, S, and Cl. A natural direction for future work is extending the model to a broader range of elements and ions, as well as integrating it with more accurate quantum-chemical methods. The model’s code has been released under an open-source license as part of the MLatom package.
The work is a collaboration between the Faculty of Chemistry at the University of Warsaw and Xiamen University. Research conducted at the University of Warsaw was funded by the Polish Ministry of Education and Science under the “Pearls of Science” program and supported by PLGrid computational resources.
Publication:
Martyka, M., Tong, X.-Y., Jankowska, J., Dral, P. O. OMNI-P2x universal neural network potential for excited-state simulations. Nature Communications (2026). DOI: 10.1038/s41467-026-71380-5